mebioda
Genome HTS in biodiversity research
High-throughput sequencing
What are your experiences with:
- Any lab work, e.g. isolating DNA? PCR?
- Any sequencing? Sanger? HTS?
- Any HTS data analysis?
- What have you learned in Genomic Architecture?
- At time of writing there are multiple technologies, broadly categorized as sequencing-by-ligation (e.g. SOLiD) and sequencing-by-synthesis (illumina, Ion Torrent, 454). Reads are getting longer on all platforms, but especially on PacBio and MinION.
- A number of vendors have created numerous platforms for specific needs and requirements, e.g. data volumes, read lengths, cost, error profile, runtime
- A fairly current review is Goodwin et al., 2016
- Illumina (below) is currently the largest platform
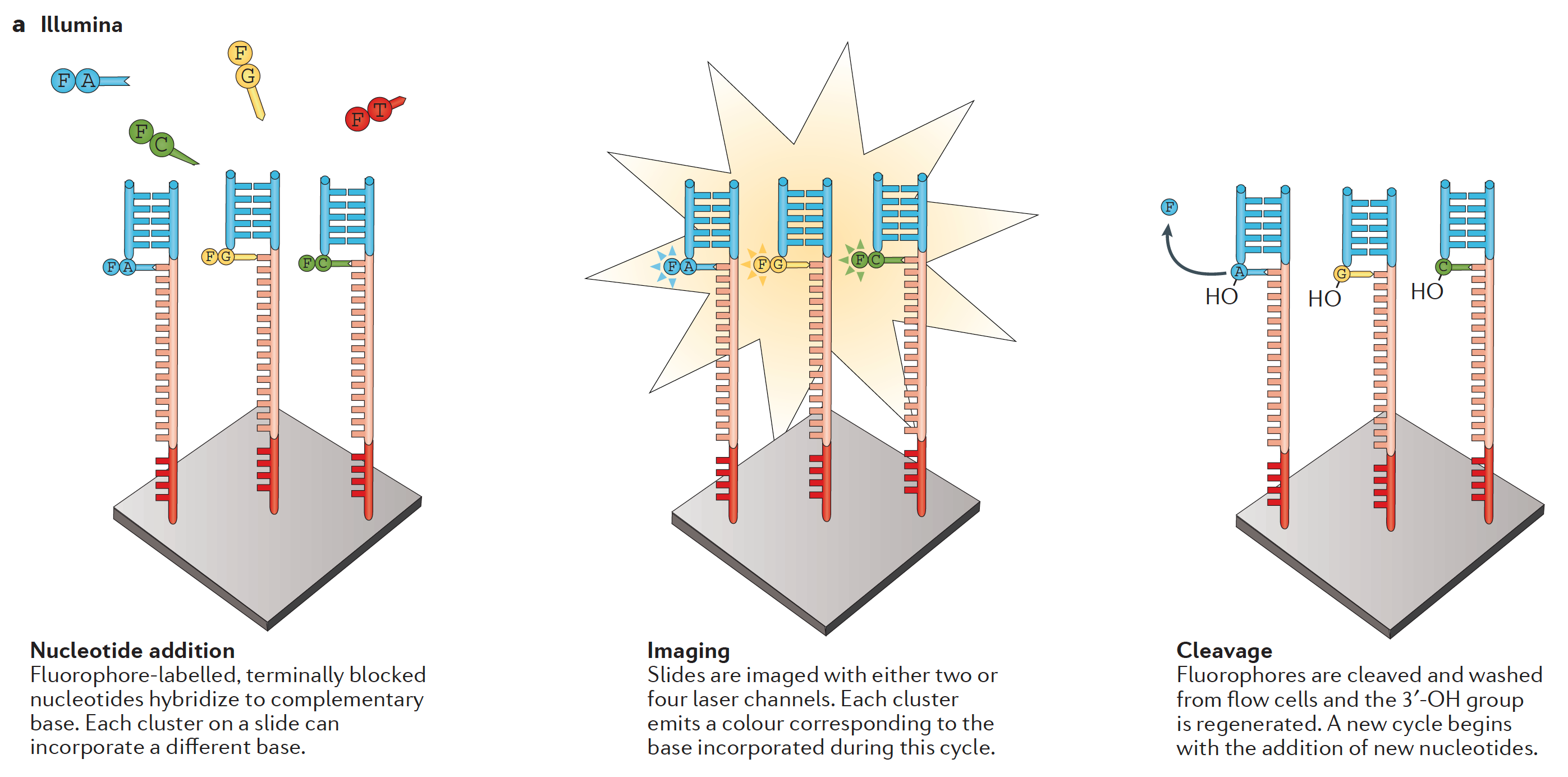
Typical workflow
- clip any synthetic oligonucleotides (adaptors, primers)
- trim low quality bases, filter short reads
- de novo or mapping assembly
- further annotation
- variant calling
- consensus sequence computation
Library preparation
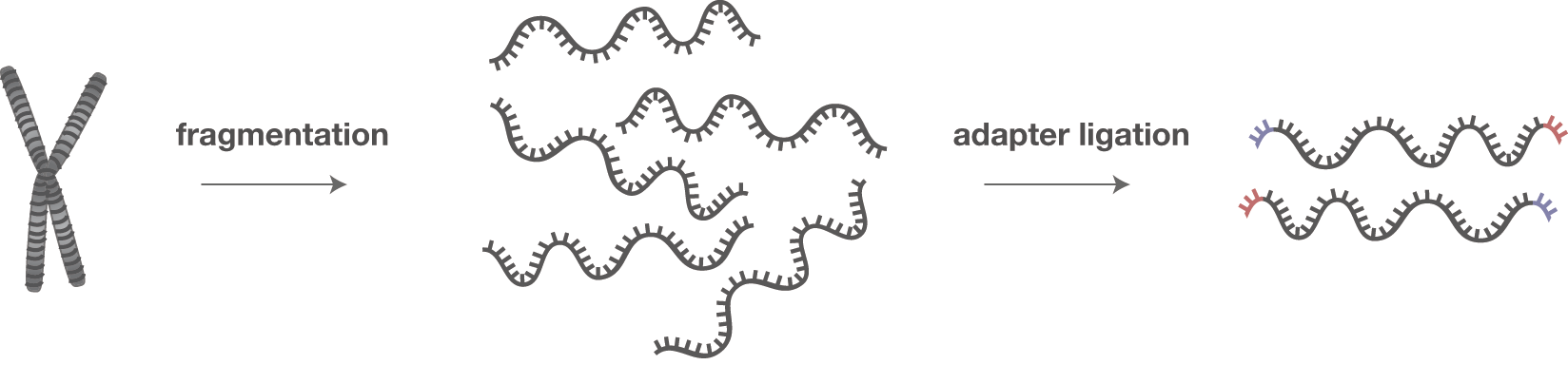
- After DNA isolation and fragmentation, primer sequences may be ligated to the fragment as part of an amplification procedure (PCR)
- In addition, various sequencing platforms involve ligation of adaptor sequences for various roles, e.g. to label samples and to participate in the chemistry of the platform (attach to the flowcell surface, for example)
Clipping adaptors
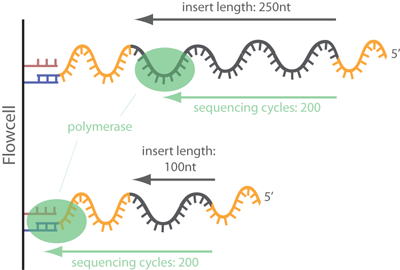
Depending on the sequencing platform, insert size, and additional services provided by the sequencing lab, the reads may already be sorted by adaptors (which are then clipped off) or you may have to do this yourself.
Effect of adaptor clipping
Sturm M, C Schroeder & P Bauer, 2016. SeqPurge: highly-sensitive adapter trimming for paired-end NGS data. BMC Bioinformatics. 17: 208. doi:10.1186/s12859-016-1069-7
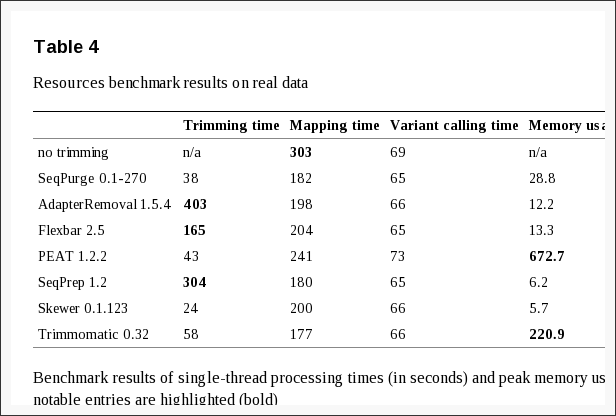
- There are many tools available for adaptor clipping. Some are faster than others, and they all affect downstream analysis time differently
- Under some circumstances, it may not be necessary to do this, depending on the experimental design (for example, if there is no de-multiplexing to do and the adaptors are ignored in a mapping assembly)
Clipping primers

Sidenote about amplicon sequencing
- In amplicon sequencing, the fragment will have been ligated with a primer as well as an adaptor sequence.
- This allows for more samples to be multiplexed because the number of combinations then becomes n adaptors * n primers
- And you probably don’t need the amount of coverage on a single marker that a non-multiplexed run would give you anyway
- However, platform vendors cannot de-multiplex automatically (because they know their own adaptors, but not your primers), and with degenerate primers you’d have to do fuzzy matching against their sequences
Quality assessment and trimming
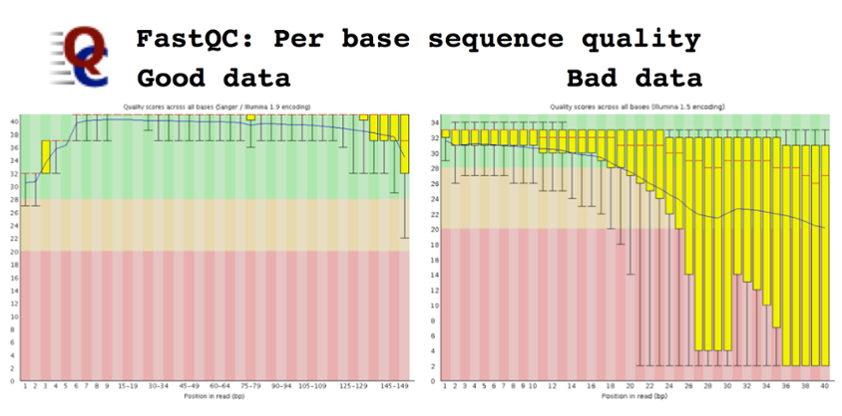
A convenient tool for initial quality assessment of HTS read data is FastQC, whose results can indicate numerous pathologies:
- Low Phred scores overall (e.g. pacbio compared to illumina), at higher base positions on the read (i.e. near the “end”), or along homopolymers, some of which can be addressed using read trimming
- The presence of biases (GC content) and overrepresentations of certain reads (e.g. adaptors) that may still need to be clipped
- Some examples show:
Additional filtering

- A (crude) proxy for a read possibly being chimeric is that it occurs only once, i.e. as a singleton, which you might therefore filter out. This is more likely the case in genomes than in amplicon sequencing (because the chimera might be PCR’ed)
- On platforms that have variable length reads, you might want to filter out all reads below a threshold length
- When paired-end sequencing, one of the two ‘ends’ might have been filtered out, in which case you might filter out the opposite end as well
Overlap-layout-consensus assembly
What do you know about graph theory? Edges? Vertices? Degrees? Directedness?
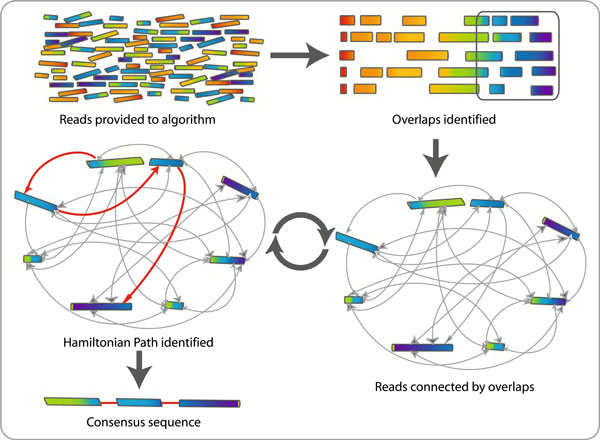
- In Sanger sequencing assembly, a graph is constructed where every vertex is a read, and edges connect the reads that overlap
- Subsequently, the Hamiltonian path, which visits every vertex (read) exactly once is searched for, and the consensus sequence along the path is computed
- However, this overlap-layout-consensus approach is hard to solve (computer scientists call this NP-complete)
An alternative way to traverse the graph
- Computing the Hamiltonian path is too computationally intensive (“NP-complete”) for HTS data
- Another approach to traverse graphs is along the Eulerian path, where every edge (instead of vertex) is visited exactly once
- The Eulerian path can be traversed in linear time (computer scientists notate this as “O(|E|)”) as opposed to non-deterministic polynomial time (i.e. with n input T(n) = O(nk) for some constant k in Big O notation)
- However, for this Eulerian path to exist, either zero or two vertices may exist with odd degree, so the Seven Bridges of Königsberg, which Euler studied, don’t form a path (and the overlap graph might also not)

Making the graph amenable to Eulerian traversal
- In a complete De Bruijn graph, all vertices have even degree
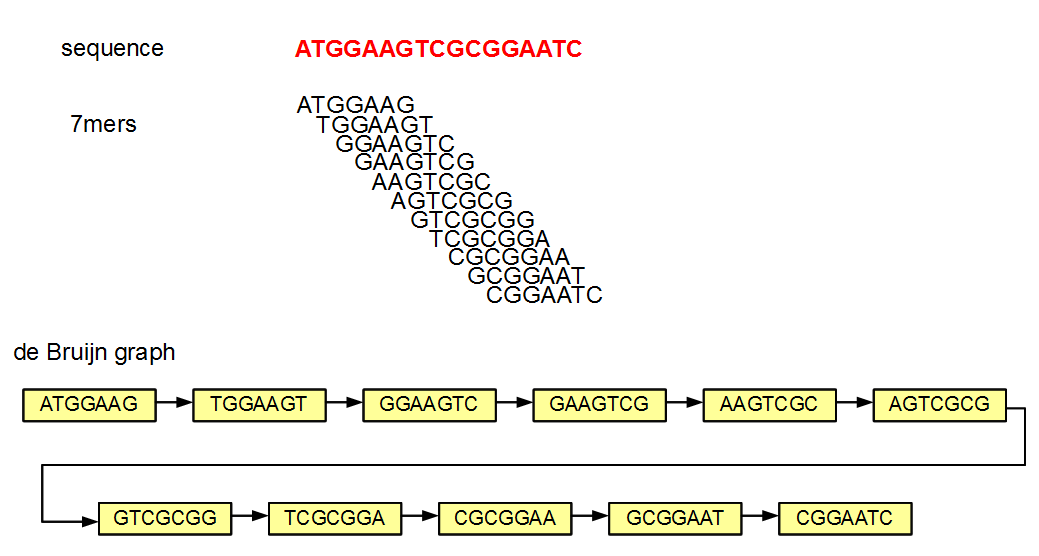
- A De Bruijn graph connects symbolic sequence data such that every vertex is a sequence string of length k (a “k-mer”) that is connected to other such vertices if the the sequences are identical along the substring of length k-1, i.e. the sequences are shifted one step relative to one another, which creates directedness in the graph
- The simplest cases, with binary sequence data, are shown for k = 1..3 (imagine what this would look like for four symbols):

HTS sequence data and k-mers
- The De Bruijn graph presupposes that for every k-mer there are neighbours that overlap with it along the substring k-1
- HTS read data does not naturally come out meeting that assumption: there are biases causing reads to overlap more irregularly
- However, the reads can be re-processed to a spectrum of substrings of some smaller size k that are shifted relative to one another (note that this collapses the duplicates that are then created):
This re-processing can be achieved naively (there are faster tools than this) in python thusly:
def find_kmers(string, k):
kmers = []
n = len(string)
for i in range(0, n-k+1):
kmers.append(string[i:i+k])
return kmers
Optimal k-mer size and assembly
Chikhi R & P Medvedev, 2014. Informed and automated k-mer size selection for genome assembly. Bioinformatics 30(1): 31–37 doi:10.1093/bioinformatics/btt310
- Smaller k makes for a smaller graph, but at the cost of more duplicate k-mers collapsed, causing information loss and an inability to cross over repeat regions
- Higher k is more memory intensive, and may increase the risk of k-mers having no neighbours, causing short contigs
- Some tools exist that attempt to optimize this value, for example by attempting to predict the value that results in the most distinct k-mers, which corresponds with certain measures of assembly quality:
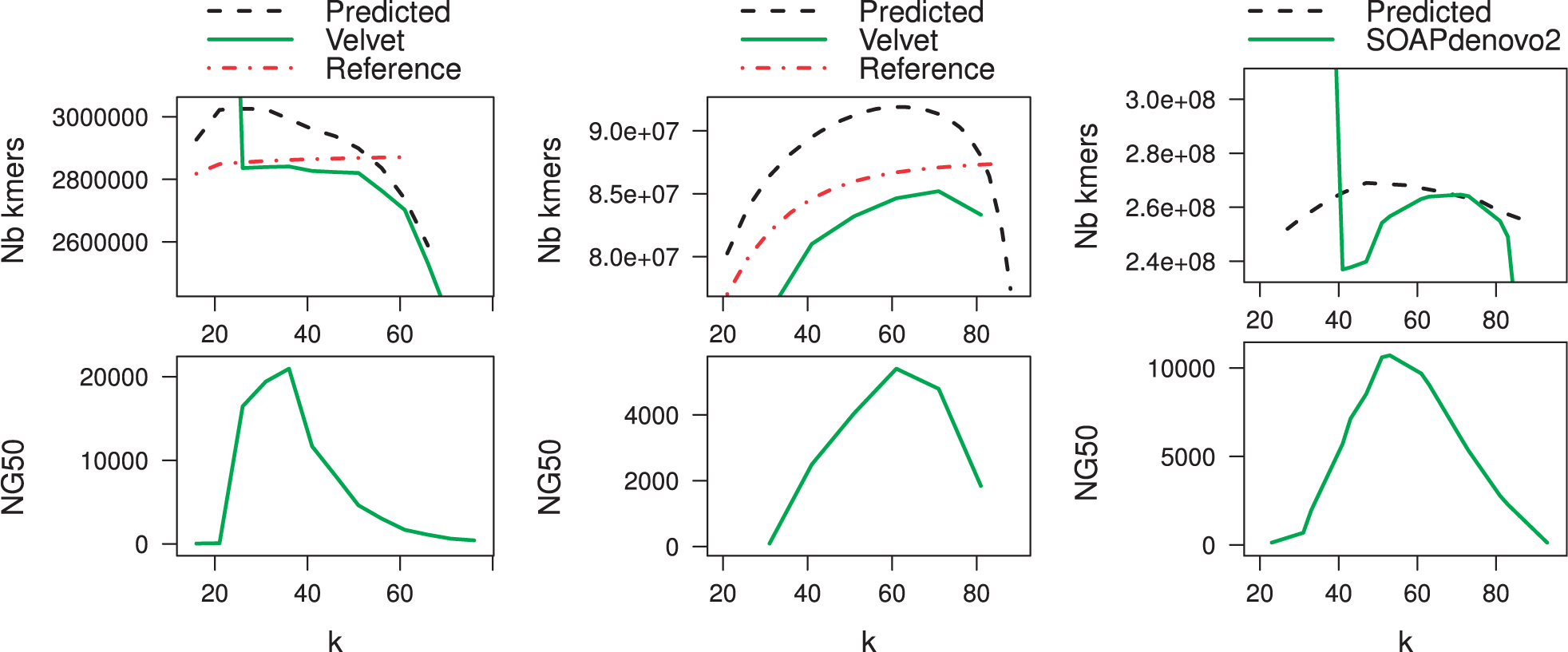
- Velvet is often used as benchmark / gold standard for de novo assembly, but it is limited in data set size.
- SOAPdenovo2 is an efficient, commonly used assembler for larger genomes.
- NG50 gives the length of the contig (when sorted by descending length) that is the mid point along the way to covering the length of the reference genome (compare with N50, which is the mid point along the way to the total length of the assembly).
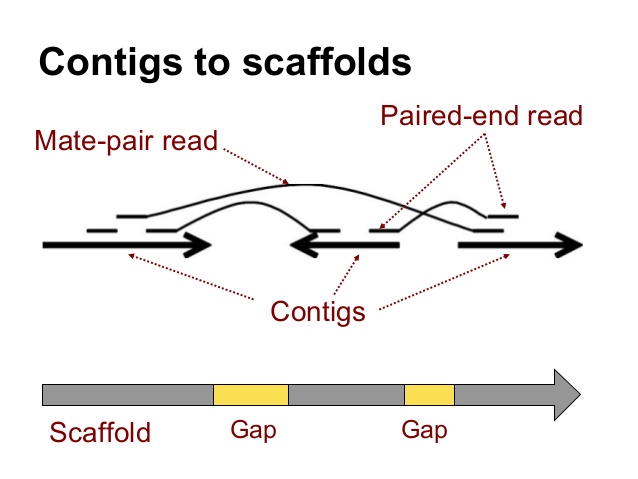
Scaffolding
- De novo assembly results in contigs, stretches of contiguous read data, which then may be scaffolded
- Paired-end and mate-pair read data (with larger insert sizes) can help span a gap and help orient and approximate the distance between reads
- Other ways to scaffold include using optical mapping or sequencing longer reads (e.g. pacbio) as well
Mapping assembly
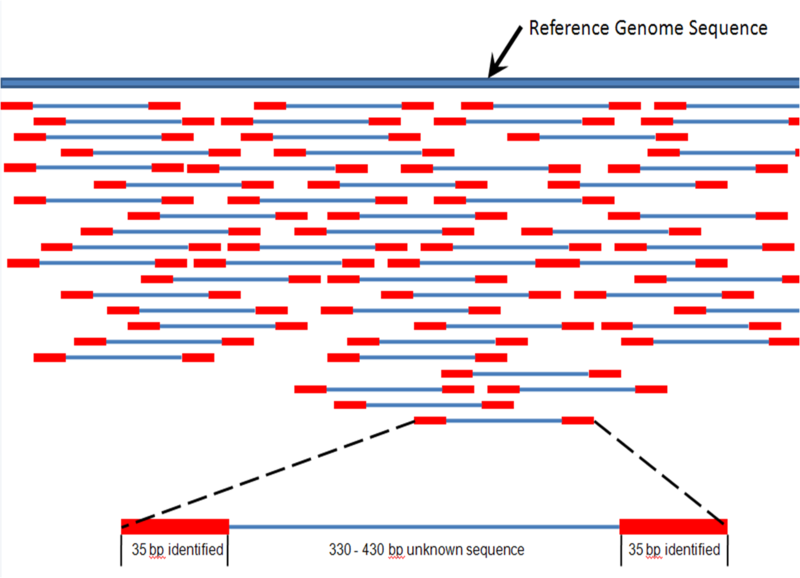
- In a mapping assembly, a reference genome is scanned for the best place to map a read
- Hence, the reference genome needs to be indexed in order for this to be efficient
The Burrows-Wheeler transform
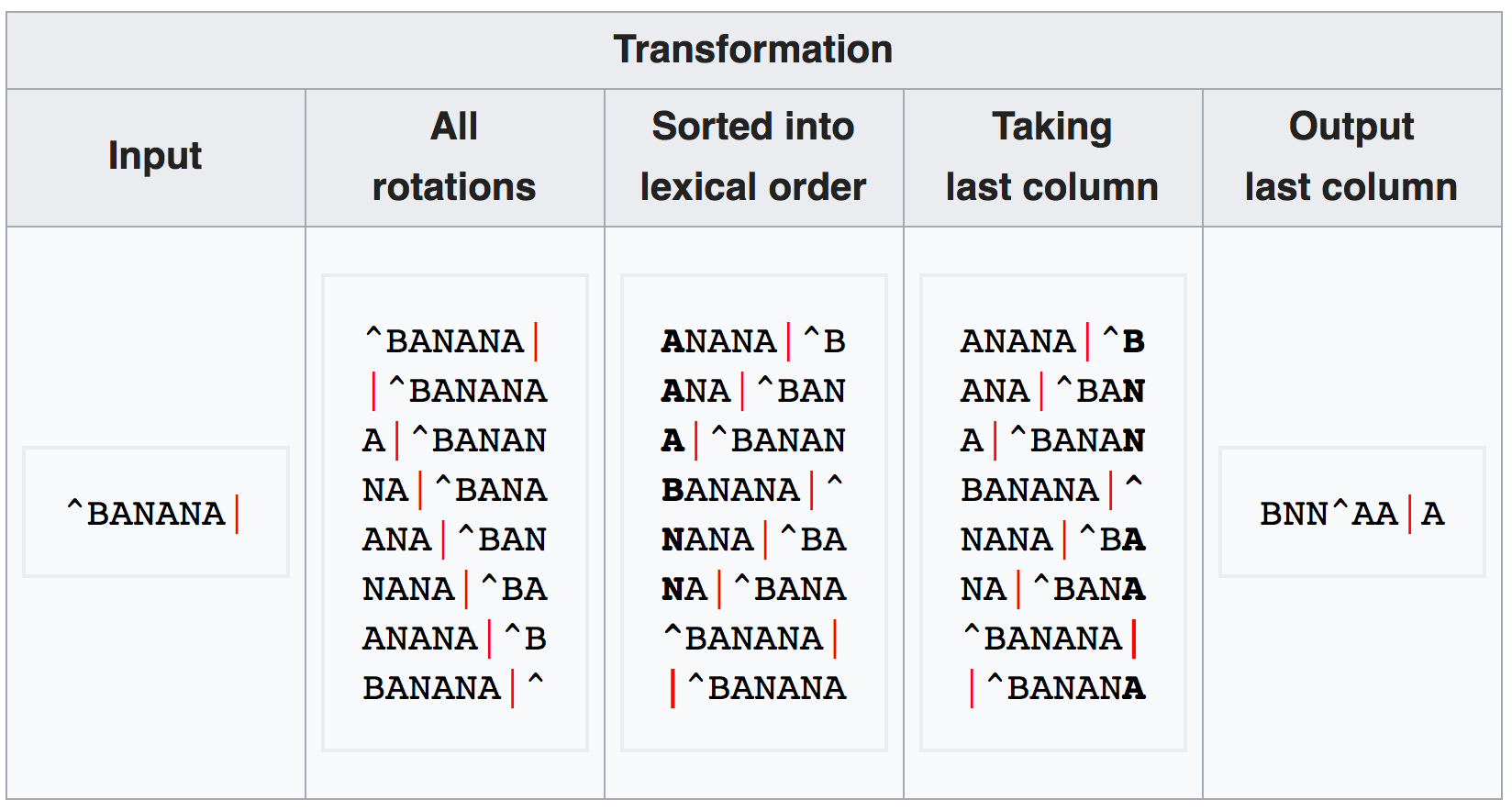
- All rotations for a given input string are generated
- These are sorted alphabetically
- The final column is the transformed string, i.e. BWT(T)
This string has the following properties:
- Almost incomprehensibly, the input string can be recovered from BWT(T) by a reverse function
- But, in the transformed string, the same character now occurs multiple times in sequence, which can be compressed (e.g. by counting the number of repetitions)
- By computing an additional index (FM), substring locations can quickly be found, and thus reads be mapped
- Here is a good tutorial with code samples for all the steps in Python
BWT mapping assembly tools
Commonly used BWT mapping tools are:
Genome annotation
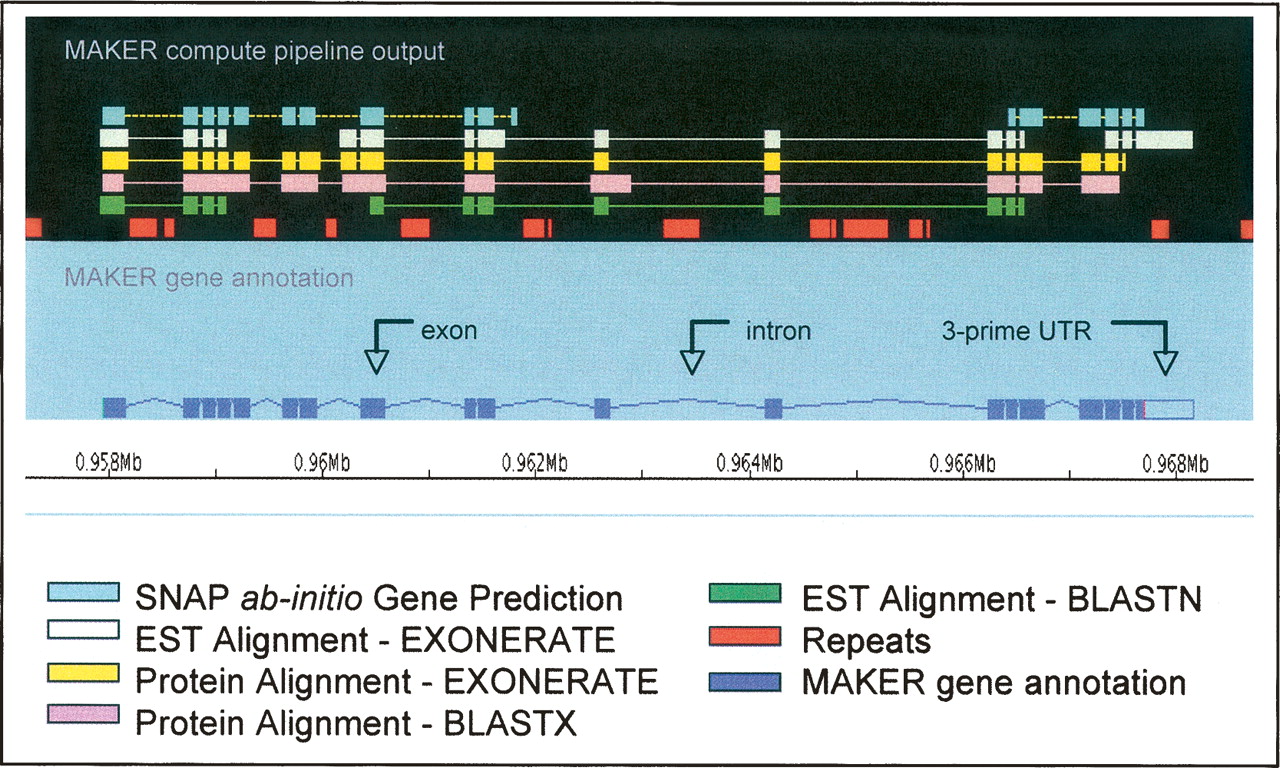
Genomes are annotated using multiple lines of evidence, such as:
- gene prediction, e.g. with SNAP
- EST mappings, e.g. with blastn or exonerate
- protein mappings, e.g. with blastx (protein blast) or exonerate
- repeat masking data
Variant calling
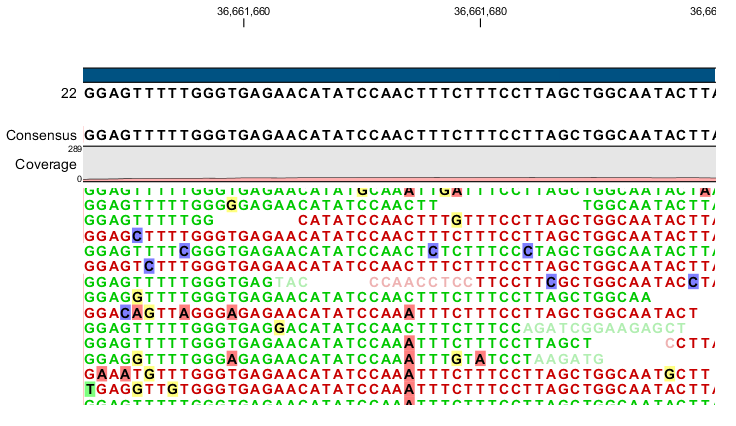
Variants are called with a variety of methods:
- Allele counting
- Probabilistic/Bayesian: build a model of the expected number of variants and use that to quantify support for any given call
- Heuristic techniques: based on filtering / thresholding for read depth, base quality, frequency