mebioda
Barcoding workflow and MSA data
Barcoding
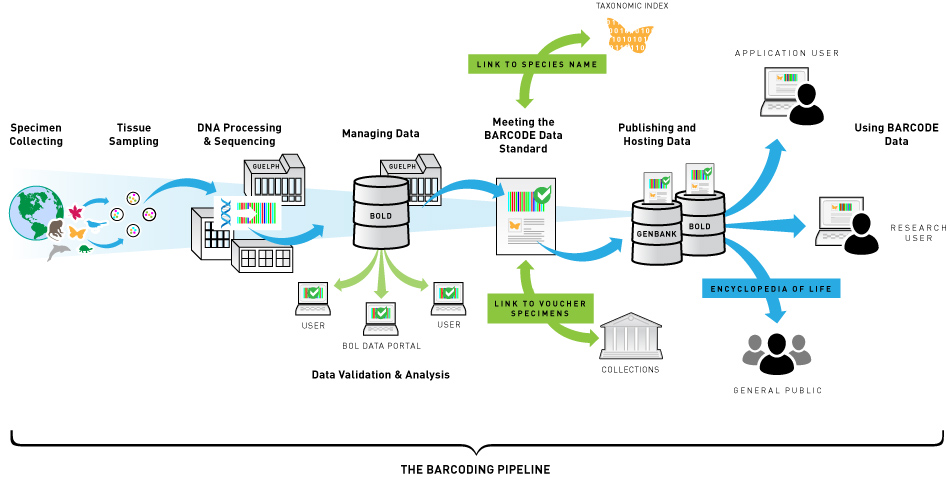
Barcode Of Life Data Systems (BOLDSYSTEMS)
- Stores records about specimens
- Includes marker sequence data, images, lat/lon coordinates, etc.
- Can query taxonomically and download all sequences
- Identification services:
- COI for animals
- ITS for fungi
- rbcL and matK for plants
Fetching taxon data through the URL API
# Fetch taxon data for Danaus as JSON
$ curl -o Danaus.json http://www.boldsystems.org/index.php/API_Tax/TaxonSearch?taxName=Danaus
Returned taxon data is encoded as JSON (e.g. do python -m json.tool Danaus.json):
{
"top_matched_names": [
{
"taxid": 6926,
"taxon": "Danaus",
"tax_rank": "genus",
"tax_division": "Animals",
"parentid": 3681,
"parentname": "Danainae",
"representitive_image": {
"image": "JAGWI/WIJAG1191+1445394342.JPG",
"apectratio": 1.333
},
"specimenrecords": "517"
}
],
"total_matched_names": 1
}
Let’s say we wanted to figure out what the image location was, we can then read this JSON in a little script:
import urllib.request
import simplejson as json # sudo pip install simplejson
url = "http://www.boldsystems.org/index.php/API_Tax/TaxonSearch?taxName=Danaus"
with urllib.request.urlopen(url) as response:
data = json.loads(response.read())
if data['top_matched_names']:
for name in data['top_matched_names']:
if name['representitive_image']:
print(name['representitive_image']['image'])
Which we run as:
python3 ../src/parse_json.py
Fetching sequences
Data from URLs can be downloaded on the command line using curl:
# Fetch all sequences for Danaus as FASTA
$ curl -o Danaus.fas http://www.boldsystems.org/index.php/API_Public/sequence?taxon=Danaus
The BOLD sequence data service API returns a FASTA file Danaus.fas, which holds
multiple sequences, unaligned. The
definition line is
formatted as:
>ID|Scientific binomial|marker|...
With this we can use standard UNIX command line tools grep, cut, sort and uniq to do some basic checks, e.g.:
$ grep '>' Danaus.fas | cut -f 3 -d '|' | sort | uniq
ArgKin
CAD
COI-3P
COI-5P
COI-5P
COII
COXIII
CYTB
EF1-alpha
GAPDH
IDH
MDH
ND1
ND2
ND3
ND4
ND4L
ND5-0
ND6
RpS2
RpS5
Wnt1
Filtering out markers
It turns out there are multiple markers for this genus. Unfortunately, because FASTA
records are multiple lines (and the exact number is unpredictable), we can’t easily use
command line tools (like grep). Instead, we might write a little script, e.g. in
biopython:
from Bio import SeqIO # sudo pip install biopython
with open("Danaus.fas", "rU") as handle:
# Example: retain COI
for record in SeqIO.parse(handle, "fasta"):
fields = record.description.split('|')
if fields[2] == 'COI-5P' and len(record.seq) == 1246:
print( '>' + record.description )
print( record.seq )
Run as:
$ python ../src/fasta.py > Danaus.COI-5P.fas
Multiple sequence alignment
FASTA files can be aligned, for example, with muscle:
$ muscle -super5 Danaus.COI-5P.fas -output Danaus.muscle.fas
Resulting in a file Danaus.muscle.fas, which is also a FASTA file.
Alternatively, you might align with mafft, (or one of the many other multiple sequence alignment tools) which has additional functionality for more difficult markers (such as ITS):
$ mafft Danaus.COI-5P.fas > Danaus.mafft.fas
Resulting in a file Danaus.mafft.fas. You can view both, for example, with mesquite.
Are they different?
$ ls -la Danaus.m*
-rw-r--r-- 1 rutger staff 157414 Dec 2 00:51 Danaus.mafft.fas
-rw-r--r-- 1 rutger staff 156819 Dec 2 00:54 Danaus.muscle.fas
Different number of bytes. Could be capitalization, could be line folding, could be sequence order, or actual differences in the alignment algorithms.
How might we verify this further?
Comparing different alignment results
Because the FASTA records in the files produced by mafft and muscle are in a different
order and the sequence data is capitalized differently, we can’t easily compare the two
files. Here we sort the records, and capitalize the sequences, then write out to a
specified file format (e.g. phylip).
import sys
from Bio import SeqIO
from Bio import AlignIO
from Bio.Align import MultipleSeqAlignment
with open(sys.argv[1], "rU") as handle:
records = {}
for seq in SeqIO.parse(handle, "fasta"):
fields = seq.description.split('|')
seq.seq = seq.seq.upper()
records[fields[0]] = seq
aln = MultipleSeqAlignment([ records[key] for key in sorted(records.keys()) ])
AlignIO.write(aln, sys.argv[2], sys.argv[3])
Usage:
$ python3 ../src/convert.py <infile> <outfile> <format>
Now we can compare the two alignments, e.g. with a diff <file1> <file2>.
They appear to be identical.
Building a phylogeny
We can build a quick phylogeny from one of the PHYLIP files using raxml, e.g.
as follows: raxmlHPC -m GTRGAMMA -n Danaus.phylo -s Danaus.mafft.phy -p 123
The resulting tree file (RAxML_bestTree.Danaus.phylo) can then be
viewed in IcyTree